
Substance Use and Misuse Overview
Additional Videos and Readings
Video– Author/Source: Pharmacodynamics and Pharmacokinetics | A rapid review
Michael Ingram, MS, MD reviews the difference between pharmacodynamics (the study of drugs and what they do to change our bodies) and pharmacokinetics (the study of what the body does to the drug after it’s taken).
Psycologist Danielle Dick explores how our genes and our environments come together across the lifespan to create multiple pathways that can influence addiction.
Video- Author/Source: Khan Academy; Reward pathway in the brain | Khan Academy
The reward pathway of the brain is connected to areas of the brain that control behavior and memory. It begins in the ventral tegmental area, where neurons release dopamine to make you feel pleasure. The brain begins to make connections between the activity and the pleasure, ensuring that we will repeat the behavior. Sometimes this pathway is helpful but other times, it can be devastating. For example, certain drugs can trigger the reward pathway and over time, an addiction can develop.
Reflection Reading #2 – Kosmyna et al., 2025 – PrePrint – Your Brain on ChatGPT: Accumulation of Cognitive Debt when Using an AI Assistant for Essay Writing Task

Chapter 11: Neuropharmacology and Substance Use
More than 275 million people worldwide have misused drugs such as heroin or cocaine at least once in 2016. Use of some of these drugs carry significant health risks, and their use has led to hundreds of thousands of deaths, making drug use one of the highest preventable causes of mortality.
For some people, drug use turns into a substance use disorder, such as addiction. These disorders are prevalent worldwide, affecting some 30 million people globally. On top of the direct health risks associated with compulsive drug use, drug addiction has a tremendous financial burden: Alcohol, tobacco, and illicit drugs cost the US more than $740 billion dollars in lost productivity, health care costs, and legal related fees and fines.
This chapter introduces you to some of the basics of neuropharmacology, the study of drugs that affect the nervous system. Drugs are defined as some chemical substance that comes from outside the body; drugs are exogenous substances that can influence a person’s physical or mental state. The field of study of the effect of drugs on the body is called pharmacodynamics, while the reverse study, the effects of the body on the drugs, is called pharmacokinetics.
11.1 Common Routes of Administration
For a drug to influence the nervous system, it has to find a way to get into the body. Drugs can be used by several routes of administration, which can be roughly divided into two main classifications: enteral, which means they have to be absorbed through the gastrointestinal tract, or parenteral, which avoids the digestive tract. Routes of administration are particularly important in substance use, because drugs that reach the brain more quickly are more likely to produce pleasurable feelings, and thus be misused.

The most common route of administration is by oral administration, by mouth. Drugs consumed orally can come in a variety of preparations, with a pill being the most common, but they can also be foods or drinks. Swallowing substances is one of the most natural processes, making oral administration one of the most convenient ways to take a drug, generally very simple requiring no additional supplies. Orally administered drugs can be designed to release over a prolonged period of time, even up to a day, which makes it a preferred route of administration for substances that need to be present for extended periods of time. The substance can also be expelled from the body through vomiting or stomach pumping in case it is suspected as causing toxicity or in overdose. Absorption of the drug into the bloodstream can also be modified by other substances, such as foods in the stomach.
One of the major considerations of oral administration is the slow onset of effect of the drug. It requires digestive processes which work on the time scale of tens of minutes to hours, making it less effective in the case of major emergencies where speed of treatment effect is the difference between life and death. It also requires the conscious action of swallowing, so oral administration will not be useful when a person is unconscious. And if a person has nausea, they may be unable to keep the drug in their system long enough for it to be digested and absorbed.
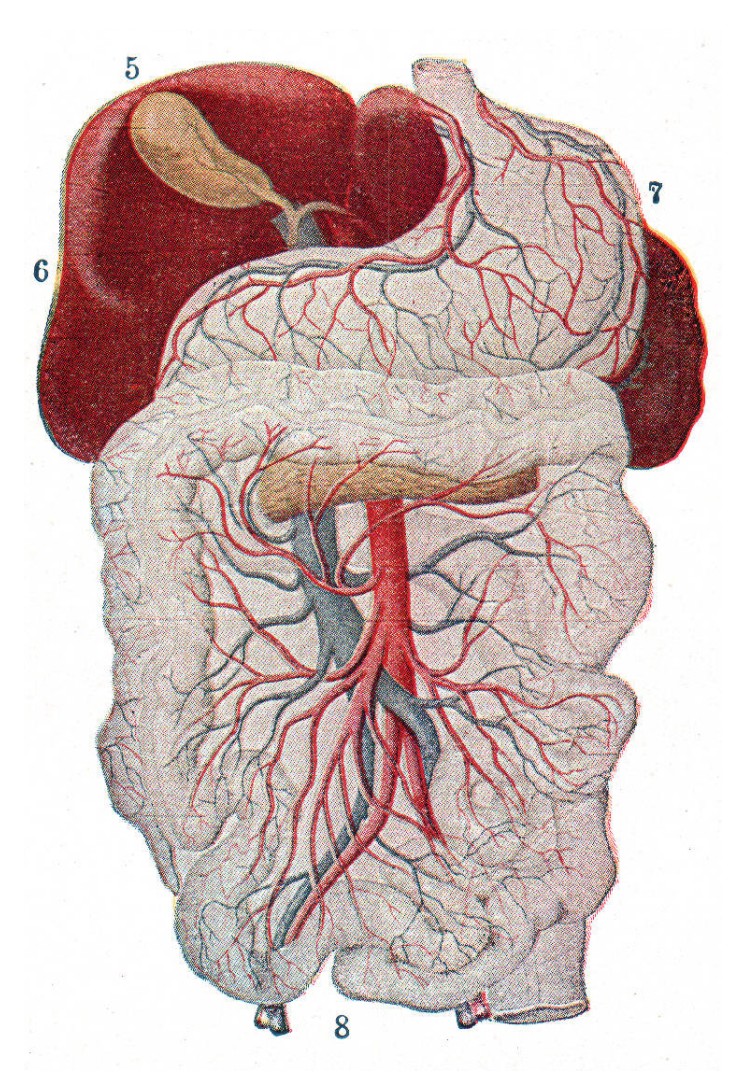
Substances that are ingested orally are absorbed through the gut wall and into the hepatic portal system, a branch of the circulatory system that surrounds the gastrointestinal tract. Before entering systemic circulation, this blood is passed through the liver, where a variety of degradation enzymes destroy some amount of the drug. This process is called first-pass metabolism, and causes a decrease in difference between the consumed amount of drug and the amount of drug that gets to its target of action.
On the contrary, some drugs are inactive until they get converted into another substance after enzymatic processing. These substances are called prodrugs. One well known prodrug is psilocybin, a chemical found in “magic mushrooms.” Psilocybin itself is inactive, but once it is exposed to highly acidic conditions, like the inside of the stomach, it becomes the psychoactive substance psilocin.
One other enteric route of administration avoids some of the issues with oral preparations. Rectal administration avoids a majority of the first-pass metabolism, and can be delivered even if a patient has severe nausea. There are two other related enteric methods worth mentioning. Sublingual refers to placement of the substance under the tongue, and may be seen with some preparations of LSD. Transbuccal administration refers to drugs that are absorbed by the vasculature in the gums as in oral nicotine preparations such as dip or Swedish snus.
Intravenous (IV) injection is the fastest route of administration, with drug effects being detected within seconds. When a needle is used to deliver the drug directly into the bloodstream, the drug is not subject to any first-pass metabolism, meaning that the dosage delivered is similar to the dosage experienced. Many substances, in particular heroin, are delivered via IV injection since it produces the most rapid onset high. In general, drugs that are taken via rapid routes of administration have a higher misuse potential. Clinically, the speed of an IV injection drug effect makes it preferable when time is essential, notably, when injecting substances like Narcan (naloxone) that can reverse the respiratory depressive symptoms of opioid overdose. Alternatively, it is also possible to deliver a pain-killer drug slowly and as needed using an IV drip, which minimizes the risks of overdose due to a sudden influx of drug.
IV injections are sort of a one-way street: the concentration of drug that enters the blood cannot be decreased. The drug rapidly mixes with blood, and can only be decreased with biological modifying agents or through natural enzymatic degradation. IV injections require syringes, which are an added cost to drug use, and bring with it a variety of health risks, including the potential transmission of bloodborne illnesses and infections at the injection site. Repeated injections can be painful and can lead to deflated veins, which is why some IV drug addicts resort to shooting drugs into other smaller veins such as the veins on their hands or between the toes.
Alternative injection routes are less common for misused drugs, and each has slightly different advantages and disadvantages. Intramuscular (IM) injections, like a flu shot into the deltoid muscle of the arm, are somewhat slower than IV injections (tens of minutes rather than tens of seconds) but are much easier to perform than an IV injection. In a subcutaneous (SC) injection, solution is injected into the fat layer underneath the skin, making it ideal for lipid-soluble drugs.
Inhalation is another rapid route of administration. Drug effects are experienced on the scale of tens of seconds. For a substance to have an effect on the brain through inhalation, it must first be volatilized then taken into the lungs through the nose or mouth. To inhale solids (such as with tobacco or marijuana), they are often burned and the smoke is inhaled, while liquids are turned into a vapor either through a heating element like vape or naturally because of the chemical properties of the liquid.
Cytochrome P450s
 In some cases, drugs interact with foods in a way that changes their effectiveness. A common example of this is the compound bergamottin, commonly found in grapefruits. Bergamottin inhibits the activity enzymes called cytochrome P450s (CYP), a class of enzymes in the liver and the small intestines that metabolize a wide variety of substances that enter the body. When we drink grapefruit juice, inhibition of CYP can decrease the dosage of drug that gets into the brain. For example, the analgesic drug codeine is a prodrug, a substance that exerts influence on the body after it is modified during metabolism. CYP normally degrades codeine into the more potent analgesic morphine, but in the presence of grapefruit juice in the body less morphine is produced, resulting in less effect than expected.
In some cases, drugs interact with foods in a way that changes their effectiveness. A common example of this is the compound bergamottin, commonly found in grapefruits. Bergamottin inhibits the activity enzymes called cytochrome P450s (CYP), a class of enzymes in the liver and the small intestines that metabolize a wide variety of substances that enter the body. When we drink grapefruit juice, inhibition of CYP can decrease the dosage of drug that gets into the brain. For example, the analgesic drug codeine is a prodrug, a substance that exerts influence on the body after it is modified during metabolism. CYP normally degrades codeine into the more potent analgesic morphine, but in the presence of grapefruit juice in the body less morphine is produced, resulting in less effect than expected.
On the other hand, other drugs are degraded by the CYP-3A4 enzyme before they are even able to affect the brain, like the anti anxiety drug buspirone. Usually, only 4% of the dosage of buspirone actually has an effect. But, when a CYP-3A4 inhibitor like grapefruit juice is present, a regular dosage of the drug results in a greatly elevated concentration in the brain, which may start to produce symptoms of overdose. More than 85 pharmaceuticals interact with grapefruit juice; some of the interactions can lead to life-threatening cardiovascular / respiratory side effects (like a buildup of the opioid fentanyl) or permanent kidney damage.
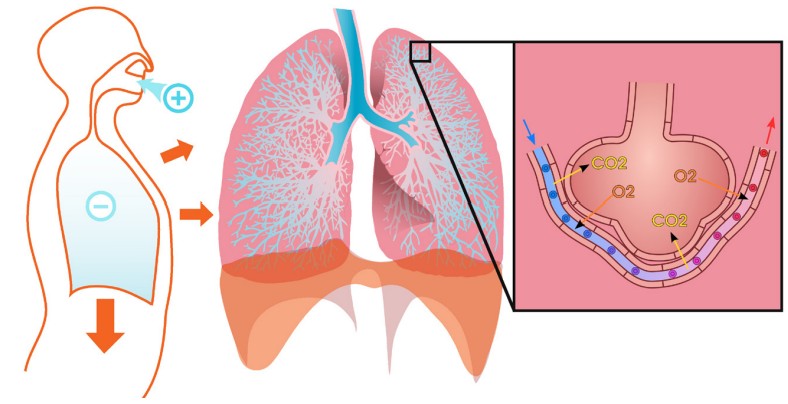
Physiologically, the lungs are extremely effective at transporting inhaled gases into the bloodstream, the same process that allows us to capture oxygen from the air and expel carbon dioxide as we exhale. Gas exchange occurs at the pulmonary capillaries, the dense network of blood vessels that surrounds the alveoli of the lungs. Inhaled air fills these alveoli and exchanges gases in the time span of each breath. This remarkable efficiency of exchanging inspired gases with the dissolved gases in the bloodstream provides one of the major clinical advantages of inhalation as a route of administration, especially during surgery. Inhalable anesthetics are very quickly cleared out of the body when a person goes from breathing the anesthesia to breathing normal air. Breathing is also one of the most natural, unconscious functions that our body possesses, which makes inhalation a very convenient route of administration.
However, inhalation of hot burnt solids causes significant damage to the airways. Inhalation of the chemicals in the propellants in compressed gases has been on the decline since manufacturers began adding bittering agents to the gases, thus making exposure to the substance highly undesirable. The dangers of this route of administration is that a person may experience hypoxia, low blood levels of oxygen, which can cause permanent brain damage. Many of the “rewarding” sensations associated with inhalant misuse are due to hypoxia.
Insufflation represents a different route of administration from inhalation because there are a few major differences of how the drug gets into the bloodstream. The substance is first pulverized into a fine powder then inhaled through the nostrils. Most of the solid sticks to the inside of the nasal cavity, where the chemical can then be absorbed by the thin blood vessels that line the inside of the nose. Insufflation, or “snorting,” is a very common route of administration for the recreational use of cocaine, producing a very rapid drug effect within minutes. Because drugs that are insufflated are protected from first-pass metabolism, they yield a higher dose that affects the brain.
Transdermal is another parenteral route of administration that avoids processing by the digestive tract. In transdermal administration, a substance is placed on the skin surface, and over time the chemical diffuses through the skin into the blood vessels. The most common and well-known example of transdermal drug administration is the nicotine patch, a nicotine replacement therapy. Transdermal patches have the advantage that the drug diffuses slowly over a prolonged period of time, up to 24 hours, minimizing the abuse potential of drugs taken in this route. However, because the skin is such an effective protective barrier against the outside world, there are major limitations to the types of drugs that can be delivered via transdermal application: only small molecules and fatty lipids that are effective at milligram concentrations can be used.
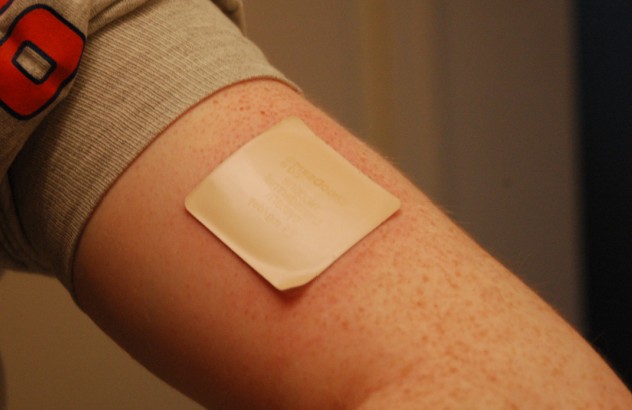
Topical administration is closely related to transdermal administration, with the major difference being that topical administration does not significantly increase the blood concentration of the drug. Topical administration acts only locally, and some common substances that are applied topically include Tiger balm or Icy-Hot. Because chemicals introduced topically do not significantly get alter brain activity, they are not psychoactive.
The pharmacokinetic profiles of drugs are important for understanding their abuse potential, and potential strategies to develop effective treatments and medications to treat overdose.
11.2 Neural Circuitry Involved in Reward
Having a sense of reward is a valuable adaptive trait. Consider two similar organisms living in the distant geologic past, both involved in Darwin’s struggle to pass their genetic material to the following generation. One of these creatures gets an internal rewarding sensation whenever it drinks water while thirsty, eats high caloric-content food, or successfully reproduces. As can be expected, this organism is highly motivated to seek out those rewarding stimuli in the world. It is are more likely to crave sugary foods to maximize daily caloric intake necessary for activity and growth. The other creature does not experience pleasure from its actions, resulting in little motivation to seek out calories or to reproduce, for example. It stands to reason that the first of these two creatures has an evolutionary advantage to survive.
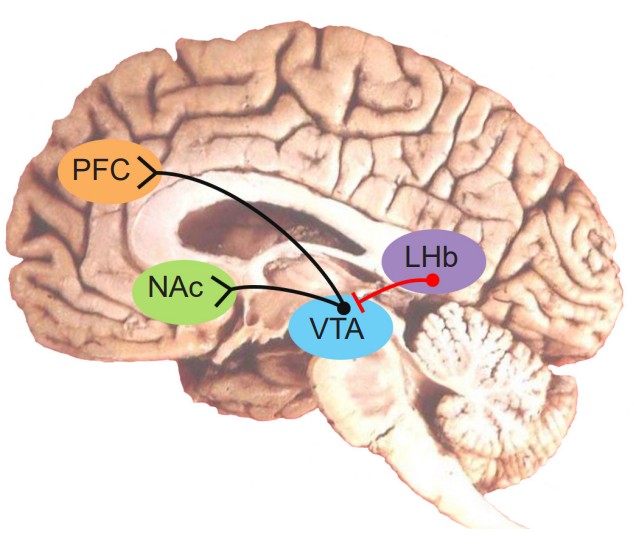
As can be expected, the regions of the brain that are concerned with reward are highly conserved through evolution. For us humans, our reward signaling neural circuitry starts with the ventral tegmental area (VTA). Located in the midbrain, the VTA contains neurons that synthesize the neurotransmitter dopamine (DA), and is the largest group of DA cells (citation) in the brain. They are also called the A10 neurons. This population of DA cells similar to the cells in the substantia nigra, the midbrain neurons that are lost in Parkinson’s disease (see chapter 10).
These VTA DA neurons send their axonal projections predominantly into two main areas. The mesolimbic pathway consists of dopamine producing neurons that release dopamine onto the cells in the nucleus accumbens (NAc, sometimes also called the ventral striatum). This seems to be the major pathway by which reward is mediated by the brain. Using microdialysis, it is possible to measure the dopamine concentration in the NAc during performance of some behavioral tasks. In studies with rats, engaging in rewarding activities such as eating or sex results in increased release of dopamine in the NAc. Similarly we see that release of dopamine into the human NAc is enhanced when we engage in activities we enjoy: binge eating sugary stuff, having sex, even playing video games. Notably, many drugs of abuse such as cocaine or amphetamine can induce a similar overflow of dopamine release when an animal is exposed to the drug.
The second area that receives VTA innervation is the prefrontal cortex (PFC), and this projection is called the mesocortical pathway. We think of the PFC as being involved in the conscious, decision making and inhibition of actions. It is likely that maladaptive changes in the dopamine projections into the PFC may explain the poor decision making and loss of control that addicts experience – their prioritization of immediate reward despite bad long term consequences (losing their job, alienating family, immediate health risks, for example). This connection has been heavily studied in the context of schizophrenia and other psychological pathologies.
Opposite of the reward pathway is a related set of neural circuits that signal an opposite reaction: aversion. If eating a sugary, chocolatey treat activates the reward circuitry, then eating a bitter-tasting bug activates the aversion circuitry. Just as with the VTA and related structures of the reward pathway, the ability to detect aversive stimuli is highly conserved throughout evolution. For example, we know that noxious plant toxins, the kinds of foods that would have made our proto-human ancestors sick, are often bitter (see Chapter 9). Being able to detect these flavors as disgusting would be a pro-survival adaptation.
If the VTA to NAc projection is the reward pathway, the projections between the neurons in the lateral habenula (LHb) to the VTA is the “antireward” pathway. The LHb sends inhibitory GABA projections onto the VTA DA neurons, so when an aversive stimulus is presented, the LHb neurons increase in their activity, which decreases DA neuron excitability through GABA release, thus resulting in less DA in the NAc. These results were conducted with electrophysiological techniques and first demonstrated in monkeys in 2009. The researchers found that when a monkey was given a squirt of delicious fruit juice, LHb neurons spiked less, which relieved the inhibition on the VTA neurons, allowing them to increase in spiking – as can be expected when a rewarding stimulus is presented. On the other hand, when an aversive stimulus was presented, in this case an air puff to the face, the LHb increased in activity while the VTA neurons decreased in activity. Relatedly, if an animal is trained to expect a reward after some innocuous stimulus (Pavlovian conditioning), and that reward is withheld after presentation of the stimulus, the LHb increases in activity.
Skinner Box
One of the earliest experiments demonstrating functional evidence of a “reward circuit” was conducted at McGill University in the early 1950’s. Drs. James Olds and Peter Milner carried out surgery on anesthetized rats to implant metal electrodes into different areas of the brain. After recovery, the rats were placed into an operant conditioning chamber, also called a Skinner box. These special cages are equipped with a device which the subject can physically manipulate. For example, the cage may have a lever which the animal can push, or a hole into which they can poke their nose. In the case of Olds and Milner’s experiments, a lever press led to activation of the implanted electrodes, resulting in neuronal activation. They had found that when the electrode was implanted into brain areas such as the corpus callosum or the hippocampus, the rats did not spend significant time pressing on the lever. However, the rats which had the electrodes implanted in the septal nucleus responded frequently on the lever. One such rat pressed the lever almost 2000 times in less than an hour, averaging nearly one response every two seconds! Electrical activation of reward centers is so desirable, rats will choose electrical stimulation over food even to the point of starvation.
This experimental paradigm is also called intracranial-self stimulation (ICSS). And while the original studies were conducted on rats, clinicians soon after tested their model in humans, with similar indwelling electrodes in different brain regions. Unsurprisingly, humans also have areas of the brain that are responsible for encoding reward, and when given the chance to electrically activate those areas, they do so extensively. When the experimenters brought in a tray of delicious food to the test chamber, the hungry patients – who hadn’t eaten for upwards of seven hours – simply looked at the meal, but couldn’t pull themselves away from the stimulation button long enough to eat. Artificial stimulation of these reward centers of the brain was more valuable to the patients than nutrition.
11.3 Molecular Pharmacodynamics
To talk more accurately about the action of drugs on a molecular level, it’s important to establish some of the vocabulary that is used in pharmacology. As we begin discussing the action and movement of molecules, it’s important to remember that the physical movement of molecules is more random than guided. When neurotransmitter molecules are released into the synapse, they move around in that space randomly. At many points in time, they float around the synapse, causing no effect. But sometimes, molecules will bump into a receptor, activating it as long as the molecule remains attached. Other times, those molecules will bump into reuptake proteins, large transmembrane proteins that decrease the concentration of neurotransmitters in the synapse by pulling the neurotransmitter back into the presynaptic axon terminal. And at other times, the molecule can even float outside of the synaptic space. Pharmacologists use the word stochastic to describe this randomness. The location of a molecule has no influence on where it will go next.
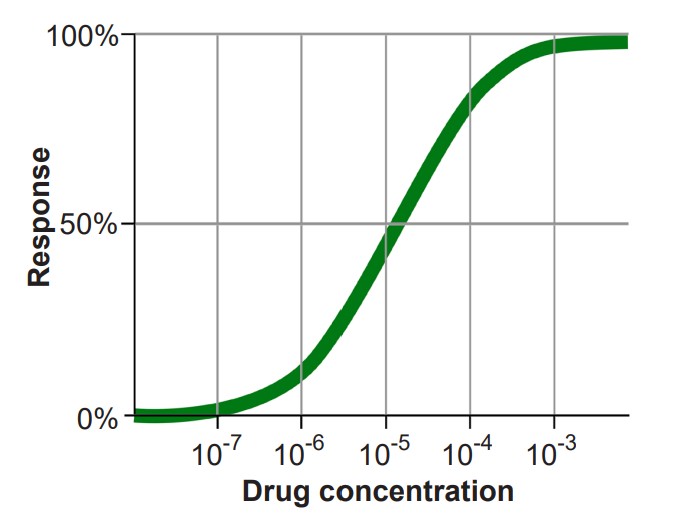
It is also helpful to become familiar with the dose-response curve, which is a graph that plots the activation of a receptor on the y-axis, and increasing dosage of a drug on the x-axis. In most cases, as neurotransmitter concentration increases, so does the activation of the corresponding receptor. The shape of the dose-response curve is described as sigmoidal, the S-shaped curve that is common to many fields of science, including ecology, microbiology, physics, and chemistry.
In molecular pharmacodynamics, we will describe three main categories of ligands, substances that are able to bind to receptors to form a ligand-receptor complex.
1. Agonists are chemical substances that can activate receptors. Receptors themselves are large, transmembrane protein structures with a surface that is exposed to the extracellular side. On this exposed side, there is some three-dimensional arrangement of amino acids that the agonist is able to bind to. When the agonist-receptor complex is formed, the receptor physically changes conformation, which can then trigger a downstream reaction. In the case of an ionotropic receptor, the protein changes to either allow or hinder ion movement across the cell membrane. Nicotine is an example of an agonist that binds to ionotropic acetylcholine receptors. For metabotropic receptors, the activated agonist-receptor complex results in downstream activation of G-proteins, which then signal the cell to behave in a variety of ways. Morphine acts as an agonist at opioid receptors, which are metabotropic. The specific site on the receptor protein where agonists bind is called the orthosteric site, or active site.
The interaction between a receptor and an agonist can be described by the analogy of a lock and key. The receptor is represented by a lock. Receptors are large protein complexes that span the cell membrane, just like a lock sits at the interface between two rooms. Receptors have an outward facing side that responds to the presence of an agonist by physically changing shape, similar to the way a lock will physically change when the correct key is inserted into the keyhole. Finally, receptors are very specific and will only respond to chemical structures that “match” the active site – the lock won’t just open in the presence of any random key.
Agonists themselves can be divided into three different classes. A full agonist is a substance that can activate the receptor to the maximal degree at high concentrations. When we think of an exogenous neurotransmitter that is released by a neuron, such as glutamate, it activates the nearby glutamate receptors maximally. In a dose-response curve, full agonists substances that set the 100% value. The phrase partial agonist is used to describe substances that can also activate the receptor by binding to the orthosteric site, but are unable to fully activate the receptor, even at increasingly higher doses. On a dose response curve, the partial agonist activates the receptor to a lesser degree than the full agonist. Some partial agonists can be used clinically for treating a variety of disorders including anxiety, psychosis, and chronic pain. Another class of agonists are the inverse agonists, which causes an opposite response as an agonist.
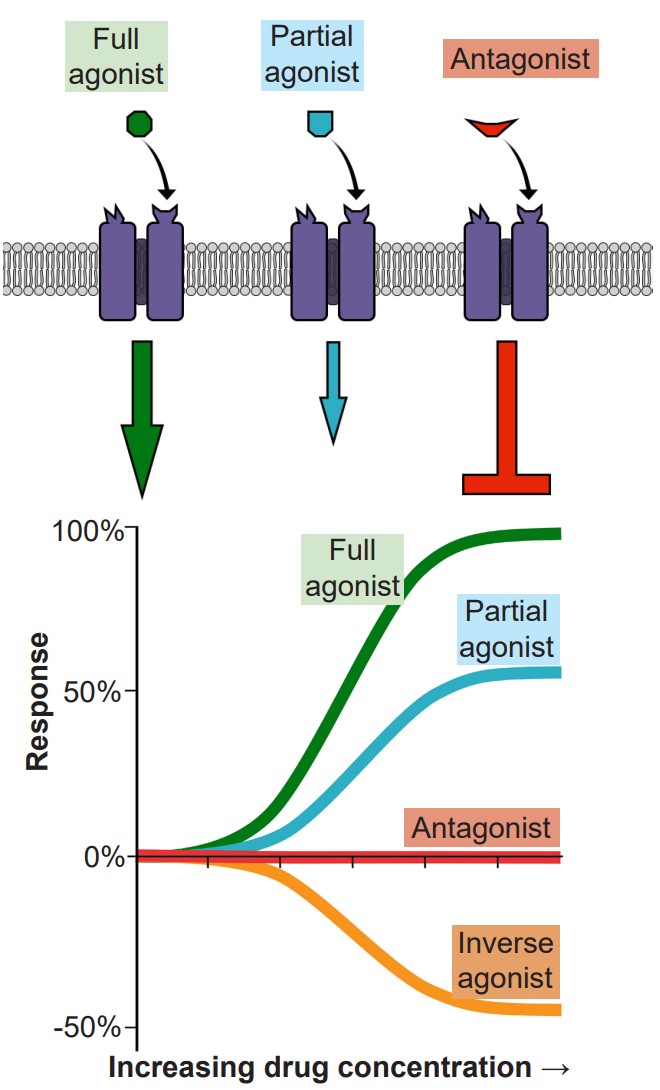
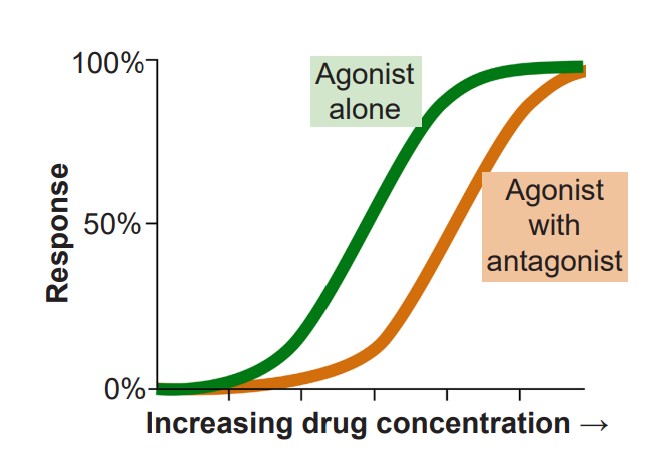
2. If agonists are the chemical substances that activate receptors, antagonists are the substances that prevent agonists from acting. More specifically, competitive antagonists are substances that bind to the orthosteric site, the same site on the receptor where an agonist would bind. Because competitive antagonists physically block the active site, and because molecules move around randomly, the presence of the antagonist can prevent the agonist from activating the receptor in a concentration dependent manner: The more antagonist present, the higher the concentration of agonist must be in the synapse for activation. Therefore, adding a competitive antagonist to the system will shift the dose response curve to the right.
One of the most well known competitive antagonists is the drug Narcan (naloxone) which inhibits certain opioid receptors. One reason opioid overdose can be fatal is the drugs are able to shut down CNS drive of the respiratory system through acting as agonists at opioid receptors. In order to treat the person in this situation, you will have to find a way to block the action of the opioid molecules that are still acting on the receptors. When Narcan is given via an IV injection or nasal spray, it works within minutes to block the action of opioid drugs that are affecting respiratory control, thus restoring normal breathing.
It is important to note that in the absence of an agonist, an antagonist alone will have no effect on cell excitation. Therefore, if an antagonist alone induces a change in some cellular property such as excitability, then it is reasonable to conclude that the cell is being acted upon by some agonist at rest.
Separate from agonists and antagonists are a different class of chemical substances called allosteric modulators that also interact with neurotransmitter signaling. These chemicals belong to a unique classification of substances distinct from agonists and antagonists. Instead of binding to the orthosteric active site, they target a different region of the receptor, a site called the allosteric site. The allosteric site generally can be found on the extracellular side of the receptor, and like the active site, consists of a special three-dimensional arrangement of amino acids. When allosteric modulators bind to the receptor, they change the potency of agonists that activate the receptor. They can either increase the action of the agonist (positive allosteric modulator, or PAM) or decrease the action (negative allosteric modulator, or NAM.) In either case, some agonist, either endogenous neurotransmitter or exogenously-introduced drug, must be present for the allosteric modulator to have any effect.
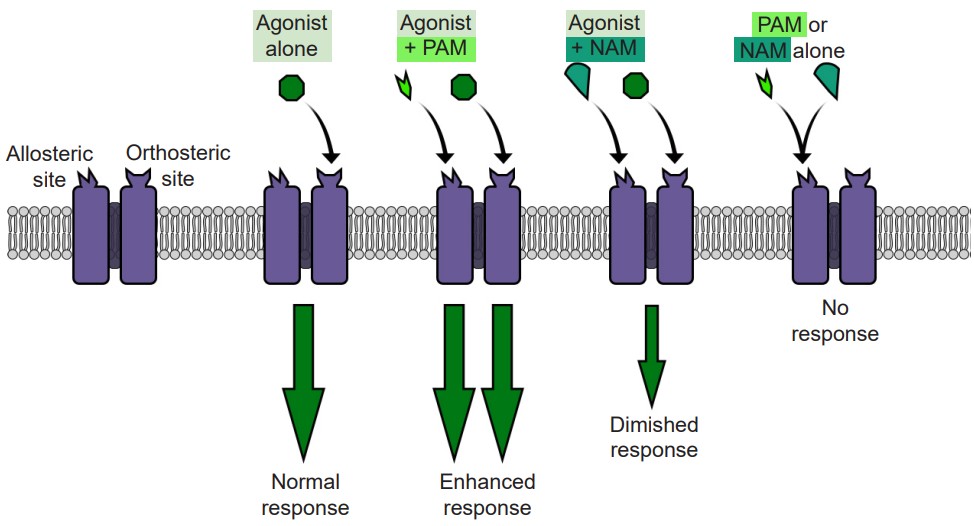
Allosteric modulators act at the level of the receptor to modulate excitability of the neurons. For example, consider an ionotropic receptor that allows Na+ to move across the cell membrane when an agonist is present. When a PAM is present simultaneously with the agonist, more Na+ ions will move across the receptor than if the agonist was acting alone. But, when a NAM is present with that same agonist, fewer Na+ ions will move across the membrane, resulting in decreased excitability.
Many of these allosteric modulators are used experimentally but some are used clinically. The most well known allosteric modulators are the barbiturates and benzodiazepines. Both are PAMs which act at the GABA receptor, meaning that when a benzodiazepine (like Valium) is present, the same amount of GABA will have a stronger effect. Since GABA is an inhibitory neurotransmitter, enhancing this inhibition of neural circuitry can be an effective way to decrease brain disorders that result from overexcitation of the brain: anxiety, epilepsy, and insomnia.
A single pharmaceutical agent can have different actions at different classes of receptors. For example, the common antipsychotic clozapine is an antagonist at some dopamine and serotonin receptors, while also acting as a partial agonist at a different population of serotonin receptors. In pharmacology, drugs with many sites of action are commonly called dirty drugs.
11.4 Commonly Misused Substances
In the early 1900s, US laws regulating the manufacture, distribution, possession, and use of drugs were complicated. Certain substances were banned, but regulation of the substances was left largely to individual states. It wasn’t until 1970 when a nationwide proposed regulatory measure would combine all of the current federal mandates regarding misused drugs into a single bill, and Richard Nixon signed the Controlled Substances Act (CSA) into law.
Under the terms of the CSA, the Drug Enforcement Agency and the Food and Drug Administration would be responsible for developing regulations about a wide variety of chemical substances. For each drug, they would be tasked with evaluating the health related properties of the drug:
1. Impact on health, including toxicity harms and addiction potential
2. Applications in the medical field

With these characteristics in mind, the substances were classified into one of five categories, rated Schedule I through Schedule V. Drugs put into Schedule I are substances that were perceived to have the highest addiction potential, most severe health risks, and no medical applications. On the other end of the spectrum are Schedule V substances, drugs with low addiction potential, few health risks, and legitimate uses within the clinical setting. Substances classified under Schedule I include heroin, for example, while a Schedule V substance might be an antiepileptic drug like Lyrica. Today, the list of controlled substances contains more than 400 chemicals. Interesting, neither tobacco or alcohol is regulated under the terms of the CSA, despite these drugs being among the most misused in the US with tremendous health risks, high addiction potential, and little medical application.
There are probably thousands and thousands of psychoactive substances, chemicals that can act on the nervous system which can induce a change in behavior or mindstate. Here, we will focus largely on six classes of drugs.
Alcohol
According to the 2015 National Survey on Drug Use and Health, alcohol is the most widely used substance among these drugs, with more than 85% of adults reporting having used alcohol at some point in their lives. Chemically speaking, many substances are classified as “alcohols,” but the most common misused is ethyl alcohol or ethanol. Ethanol is the easily-obtained substance with a highly intoxicating effect. The overwhelming majority of ethanol use is recreational. The World Health Organization reports that alcohol use is the leading risk factor for premature death among males aged 15-59 globally, making it one of the overall top causes of preventable death.

Acute exposure to ethanol has effects that differ depending on dosage. At low concentrations, ethanol can induce an elevation in mood, decreased anxiety, increased risk taking, slowing of reflexes, and impaired judgment. At high concentrations, ethanol can cause memory deficits, loss of consciousness, analgesia and areflexia, and possible death through respiratory depression. Chronic exposure to low levels of alcohol has some pro-health benefits such as decreased risk of death by cardiovascular events, but in general, these benefits are outweighed by negative health outcomes, such as the elevated risks of liver disease or cancer and alcohol-related violence or injuries.
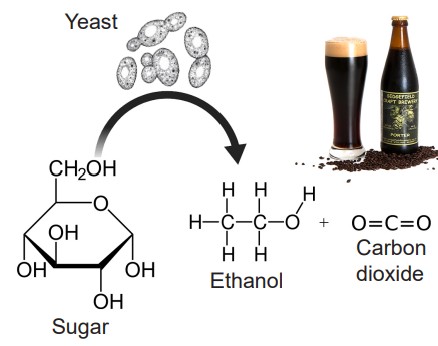
Ethanol is a metabolic byproduct synthesized by yeast as they break down sugars. It has the chemical structure of C2H6O, making ethanol a tiny molecule with properties similar to water, meaning that ethanol can diffuse easily through cell membranes. Whereas other chemical substances often have highly specific molecular or protein targets, ethanol has several: GABA receptors, glutamate receptors, potassium channels, serotonin receptors, and more. In general, ethanol has a depressant effect on neurons, but it has been demonstrated to activate DA neurons in the VTA.
Nicotine
Nicotine is the main psychoactive ingredient of tobacco products. Nicotine is an alkaloid compound that is synthesized in nature by a variety of plants, with the tobacco plant having the highest nicotine content. Other plants of the Solanaceae family (Nightshade family), such as eggplants and tomatoes, also contains nicotine, as it was probably an anti-herbivory evolutionary adaptation. The most common routes of administration for nicotine include inhalation (cigarettes, cigars, vape), transbuccal (chew, dip, snuff), or transdermal (in the case of the nicotine replacement therapeutic strategy, the nicotine patch). Nicotine is a highly addictive substance, often rated as addictive as cocaine or heroin. It is currently the leading cause of preventable death in the US, causing nearly one out of every five deaths.
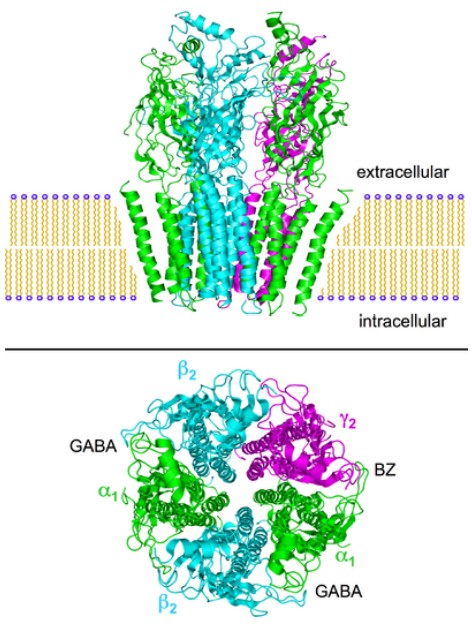
Pharmacologically, nicotine has specificity as an agonist at nicotinic acetylcholine receptors (nAChRs). These receptors are excitatory ionotropic receptors, and are expressed widely across the body. Nicotine is a stimulant, and induces release of norepinephrine by activating the sympathetic nervous system. When a person smokes a cigarette, they experience a rapid rush or a high within tens of seconds. Chronic smoking greatly increases the risk of developing lung, throat, and mouth cancers, coronary heart disease, or stroke.
Cigarette smoking over the past several decades has seen a sharp decline, in part due to the success of anti-smoking campaigns in changing public opinion toward smoking, and government-led changes in legislation resulting in stricter marketing laws, especially in regards to targeted advertising aimed at youth. In the mid 1970’s, almost 30% of 12th graders reported smoking cigarettes daily, and that number has decreased to only 4% in 2018. On the other hand, the rising popularity of vape among teenagers has encouraged nicotine consumption through an alternate route of administration: Teen e-cigarette users are more than 3 times more likely to start smoking cigarettes compared to non e-cigarette users.

Cannabis
Cannabis is a drug that is derived from the flowering buds or other parts of the Cannabis sativa plant. The most common route of administration is inhalation, as when people smoke a joint or a blunt. As cannabis becomes decriminalized or outright legalized across the US, oral preparations are becoming more commonplace. The effects of cannabis are often felt within minutes, and can include a sense of euphoria, relaxation, distortion in the sense of perception, and potentially a lightness (high) or a heaviness of body (stoned).

Although the cannabis plant contains several chemical substances, the main psychoactive ingredient is delta-9-tetrohydrocannabinol, or THC for short. THC, once it enters the bloodstream, is capable of activating receptors that our body uses endogenously, called the cannabinoid receptors (CB receptors). There are two types of CB receptors, with CB1 exerting the majority of the psychoactive effect and CB2 expressed mostly throughout the immune system. Our bodies naturally produce substances that activate these receptors; these substances are called the endocannabinoids, or eCBs. Activation of the CB1 receptors, either through eCBs or THC, produces the elevation of mood.
Cannabis is currently classified as a Schedule I drug federally, but as of 2019, 10 states have legalized recreational use of cannabis, and 20 other states permit cannabis for medicinal use. Recently, Illinois has permitted the 55 medical dispensaries to sell recreational cannabis starting in January, 2020. The stigma surrounding the “evils” of cannabis use are largely a remnant of a morality propaganda campaign driven by the publishing of such “educational” films as Reefer Madness in 1936. Much of our current medical understanding of cannabis is that it is far less harmful than once believed, although it does have a negative impact on the developing brain, as significant adolescent exposure to cannabis increases the likelihood of developing psychiatric conditions such as schizophrenia later in life.
Opioids
Opioids are a class of drugs, either natural or synthetic, that can bind to and activate the body’s endogenous complement of opioid receptors. The most well known natural opioid is opium, but the other popular drugs of this class include the street drug heroin or the medicinal substances morphine or fentanyl. Derived from the poppy plant, evidence of human use of opioids for both medical and recreational use dates back more than 5000 years to ancient Sumer.
As with the endocannabinoids described above, the body naturally produces opioid substances, endorphin being the most wellknown. Activation of opioid receptors, either by endogenously produced opioids or exogenous opioids, induce a potent analgesia, dampening the sensation of pain. Opioid drugs the current gold standard for pain relief. They also cause sedation and feelings of euphoria. Heroin and morphine are frequently taken via IV injection, but many prescription opioids, particularly for long-term pain conditions, are consumed orally.
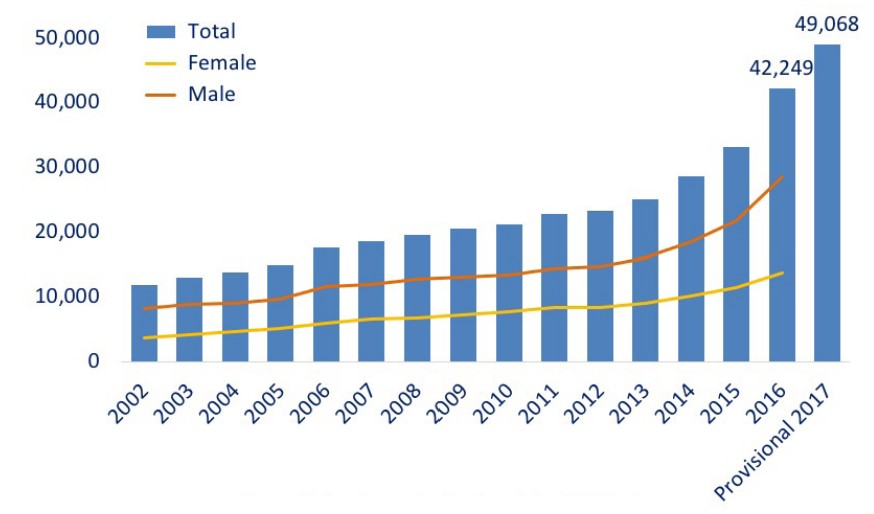
Over the past 20 years, there has been a dramatic increase in the number of opioid overdose related deaths. The most recent spike in deaths is due to overdose on synthetic opioids such as the extremely potent painkiller fentanyl, which saw a 10-fold increase between 2013 and 2017. The so-called opioid epidemic kills an estimated 130 Americans each day.
Cocaine
Cocaine is a psychostimulant that is derived from the coca plant. The most common routes of administration are insufflation (powder cocaine), inhalation (crack cocaine), but it can also be solubilized and injected or taken via transbuccal and oral administration by chewing the leaves of the plant.
Cocaine is considered to be a potent sympathomimetic, meaning the substance activates the sympathetic nervous system. Therefore, the physiology of a person on cocaine will be similar to an activated “fight-or-flight” response: elevated heart rate and blood pressure, dilated pupils, and increased respiration.
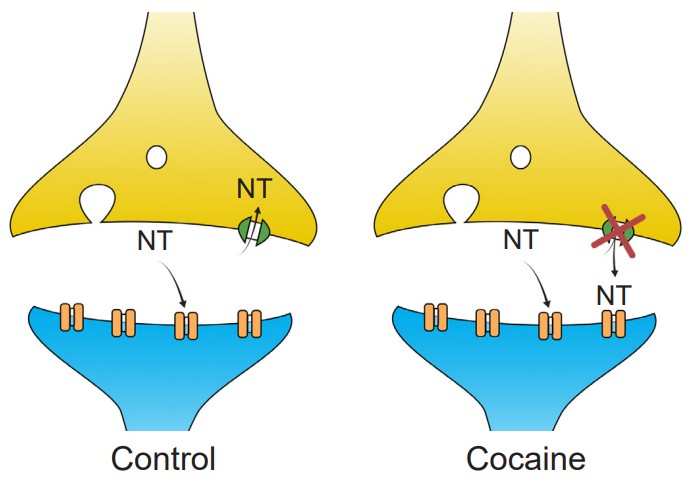
Cocaine acts on a molecular level by functioning as an inhibitor of reuptake proteins. At the synapse, there are often presynaptically expressed proteins called transporters. These large membrane-spanning proteins function to move molecules from the extracellular space back into the presynaptic terminal, where they are then repackaged into vesicles for release at a later time. Cocaine is a reuptake inhibitor, physically preventing the transporter protein from clearing the neurotransmitters dopamine, norepinephrine, and serotonin out of the synapse. This leads to an elevation of the concentration of those neurotransmitters, which results in a greater likelihood that the synaptic receptors get activated.
Cocaine is a Schedule II substance. Although it has a very high addiction and harm potential, it has a handful of medicinal applications as well. For example, it is a potent local anesthetic and vasoconstrictor, so it is often used in surgery, especially in facial or nose surgeries.
Psychedelics
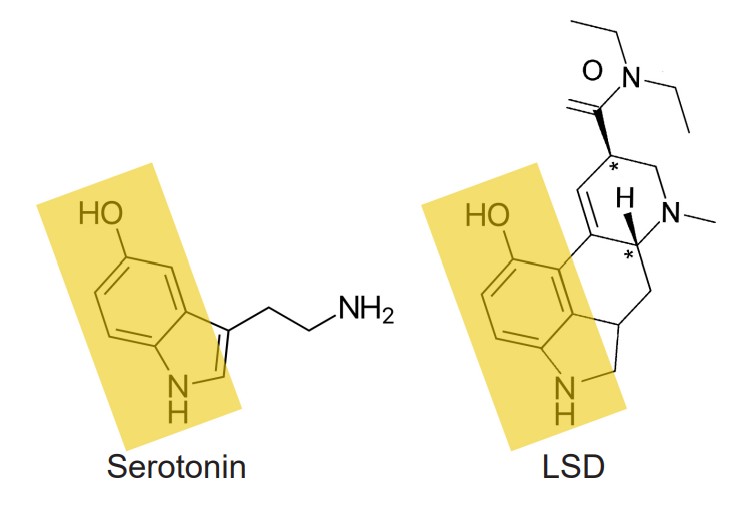
Psychedelics are the class of drugs formerly called hallucinogens. They can be either natural, such as psilocybin which is derived from mushrooms, or man-made, in the case of lysergic acid diethylamide (LSD). Psychedelics can cause a person to experience visual distortions, synesthesia, an altered sense of self (ego dissolution), or sometimes a sudden connection with nature or a higher power. Chemically, many psychedelics are similar in structure to the endogenous neurotransmitter serotonin and therefore activate serotonin receptors.
Although psychedelics have been used for centuries in religious ceremonies, they have had a troubled history in the US. The adoption of LSD (acid) by the counterculture movement during the 1970’s, secret CIA funded research, and some loosely-regulated research ethics in the clinical setting all led to notoriously negative press regarding the substance, tainted the general public opinion about this class of drugs. Since 2010, there has been a growing body of evidence supporting the notion that psychedelics can serve a therapeutic purpose, most notably in treating psychiatric conditions such as PTSD, terminal illness-related depression, and drug addiction.
11.5 Tolerance, Withdrawal, and Dependence
Pharmacologically speaking, tolerance can be defined as a decrease in the action of a drug due to repeat exposure. This phenomenon happens as a result of homeostatic adaptations that the body undergoes when it has been exposed to a substance for a prolonged period of time. In a person who is tolerant, they must take progressively higher doses of the drug to experience a desired effect.
Tolerance can be represented graphically by a rightward shift in the dose response curve. For a drug naive person, someone with no experience with the drug, a low dose of drug may have a strong effect. However, for a person who has become tolerant to the effects of the drug, that same low dose will produce a small drug effect. (The other way to think about the dose response curve is the following: For two people to experience the same magnitude of effect, a drug tolerant person would require a higher dose than someone who has never taken the drug before.)
There are different forms of tolerance that a person can experience.
In metabolic tolerance (sometimes also called dispositional tolerance, or pharmacokinetic tolerance), there is a decrease in the amount of the drug that gets to the site of action. The body becomes more efficient at eliminating the substance. Since exogenous substances usually get degraded by enzymatic reactions, an increase in the activity or number of enzymes will decrease the amount of the substance that is capable of acting.
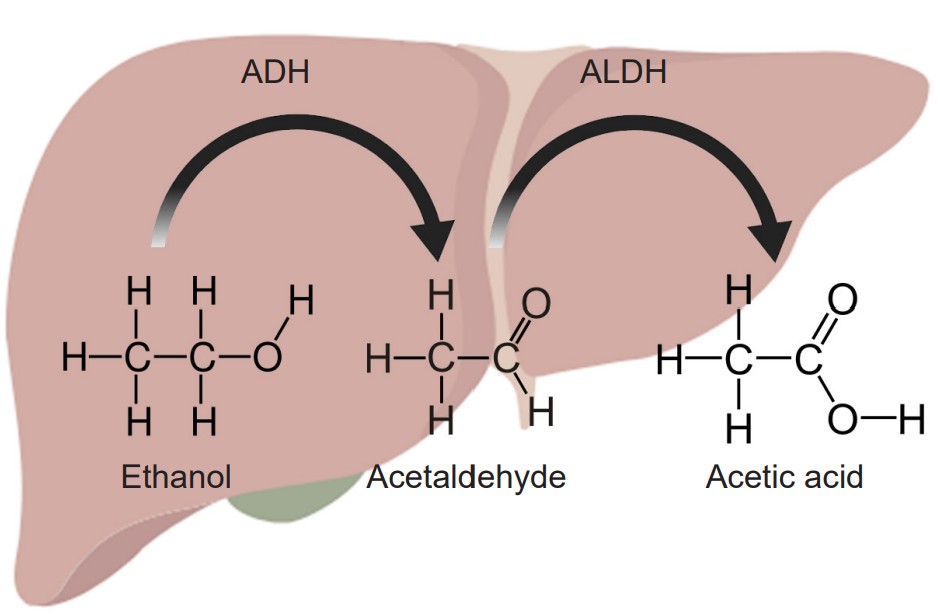
The most common example of metabolic tolerance is seen in frequent alcohol use. The main psychoactive substance found in alcoholic beverages like beer, wine, or liquor is ethanol. Our liver, which filters circulating blood, contains enzymes which break down chemicals in the blood. Specifically, the standard degradation process of ethanol uses two enzymes. The first of which, called alcohol dehydrogenase, oxidizes molecules of ethanol into the toxic byproduct acetaldehyde. The second step uses the enzyme acetaldehyde dehydrogenase to convert the acetaldehyde into acetic acid, a generally harmless compound (the same chemical structure as vinegar) that is used by the body in a biochemical reaction that drives the citric acid cycle. Because most alcohol is taken via oral consumption, ethanol is subject to first-pass metabolism before reaching systemic circulation.
However, after prolonged exposure to alcohol, the body undergoes homeostatic compensatory changes. The two liver enzymes responsible for degradation of ethanol increase in either function or amount. When these metabolic enzymes become upregulated, the circulating ethanol in the bloodstream becomes broken down more rapidly and more efficiently, resulting in a decrease in the amount of ethanol that reaches general circulation, and consequently, the brain. This explains why people who have never drank alcohol (low amounts / function of enzymes) may experience a stronger drug effect compared to someone who drinks regularly. It also accounts for why there is some genetic variation between a person’s sensitivity to alcohol’s intoxicating effect, as enzyme efficiency is partially encoded by genetics.
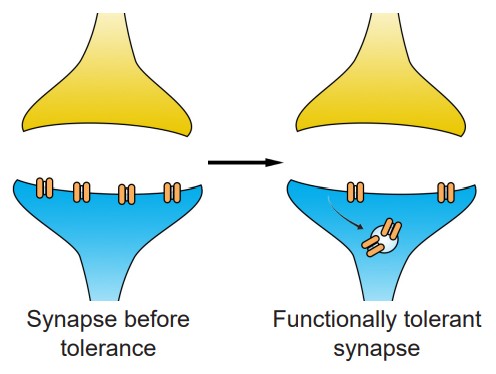
Another way by which tolerance can manifest itself is through changes at the level of the molecular components of cells. This type of tolerance is called a functional tolerance, or pharmacodynamic tolerance. This tolerance leads to a decreased sensitivity to the substance. This functional tolerance can be characterized by a decrease in receptor expression after chronic exposure to an agonist. When the opioid etorphine is present in the body, it activates the opioid receptors. Frequent exposure to etorphine causes a molecular change in the receptors, which leads to internalization, the process by which cell surface-expressed receptors become taken into the cell. Since etorphine is only able to activate receptors on the surface, these internalized receptors diminish the strength of the etorphine signal, which manifests as tolerance. By downregulating the number of receptors that are expressed on the surface, the cell reacts to the environment by becoming less responsive in the face of drug.
Sometimes, the body physiologically prepares for the drug effect before the drug is even present. The way it does this is to initiate an opposite somatic effect from what the drug is expected to do. This “reverse” effect is likely a protective mechanism to minimize the drug effect, and these anticipatory bodily changes can be precipitated by drug-associated cues. For example, heroin causes analgesia. Cues that predict administration of the drug, such as preparation for an intravenous injection or being in a physical environment where drug is frequently administered, can cause the reverse physiological effect, hyperalgesia. Because of this, a person may need to take more drug to overcome the anticipatory changes. This unconscious form of learning leads to a form of tolerance that is dependent on Pavolvian cues; this is called conditional tolerance.
If not accounted for, conditioned tolerance can have potentially lethal side effects. A clinical description (Siegel, 2001) of a patient with pancreatic cancer describes one such “failure” of tolerance. Bedridden and suffering from tremendous pain, a man was regularly given morphine in his bedroom, dark and filled with humming apparatus for his medical care. As expected, he developed a tolerance towards the morphine, requiring higher and higher doses in order to inhibit pain. One day, he went into the brightly lit living room to receive his normal dose of painkiller. Soon after, he died from opioid overdose.
Sensitization is a phenomenon related to tolerance, but the polar opposite: instead of a drug effect being lessened after chronic exposure, a sensitized person experiences an increase in drug effect. It is sometimes called “reverse tolerance.” Nicotine as well as many psychostimulants such as cocaine or amphetamine can produce psychomotor sensitization after repeated dosing.
Since mechanisms of tolerance result in long lasting homeostatic changes, they may lead to withdrawal, a set of symptoms that a person experiences when they are abstinent from the substance. This produces a highly aversive state that can be relieved by administration of more drug. The symptoms of withdrawal are frequently the opposite of the side effects of the drug. One example is the physiological response to heroin. When heroin is present, a person may experience euphoria, analgesia, relaxation, and constipation. But after the homeostatic changes have taken place resulting in downregulation of opioid receptors, a person may experience withdrawal symptoms such as dysphoria (depression), pain hypersensitivity, restlessness, and diarrhea.
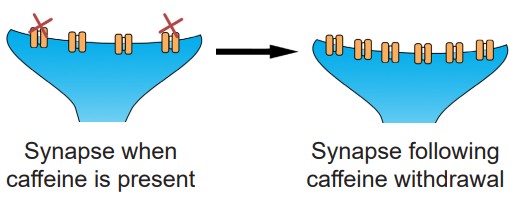
It is good to note that tolerance does not only happen in the presence of agonists like ethanol or morphine. In fact, antagonists are well known to produce tolerance and withdrawal. If you have ever had difficulty staying alert a headache or without your morning coffee, you are experiencing the effects of withdrawal. Caffeine is an antagonist for the adenosine receptors. When the body is used to a certain amount of caffeine that blocks adenosine signaling, there are mechanisms that lead to homeostatic upregulation of adenosine receptors. And so when caffeine is no longer present, the endogenous levels of adenosine overactivate adenosine signaling, leading to the withdrawal symptoms.
When someone is withdrawing from a drug, they often feel the urge to take the drug. This is a sign of drug dependence. A dependence can be a physical dependence, where the person seeks drug in order to either relieve the physical symptoms of withdrawal, or to experience the positive sensations associated with the drug. Alternatively, a psychological dependence can also develop, where the person has intense cravings for the drug such as a fixation on drug acquisition, or mood or behavioral changes in the absence of drug.
Long term drug use may cause people to become heavily dependent. These people often engage in recurring patterns of drug seeking and drug taking. Their patterns of use can be highly destructive, as they make drug acquisition as their main objective, fixating on drug taking even at the cost of their jobs, relationships, and even their own health. Because this form of pathological drug use is extremely harmful, much attention is focused on understanding the mechanisms that underlie this condition.
11.6 Theories of Addiction
“Why do some people transition from user to addict?”
Many people are able to use the most addictive substances such as cocaine or heroin casually without ever becoming an addict. For these people, their drug use pattern can remain recreational, even though they may use the substance frequently and report a great sense of satisfaction when they use the drug. On the other hand, there are some people who have a very high propensity to quickly becoming a pathological addict. One of the big challenges in drug abuse research is to understand the differences between these two populations – what factors, either environmental or genetic, are protective and prevent the transition from casual drug user to compulsive drug addict? And what are some ways we can use this knowledge to help addicts find ways to break their pattern of harmful compulsive drug seeking and drug use?
One possible way to get at the answer is to create animal models of drug dependence and try to determine the nature of the underlying biological changes that contribute to the addiction-like pattern: Do some cellular circuits change permanently? Are certain genes or proteins upregulated or downregulated? Can pharmacological or behavioral interventions change drug-taking or drug-seeking behavior?
Non-human models of addiction
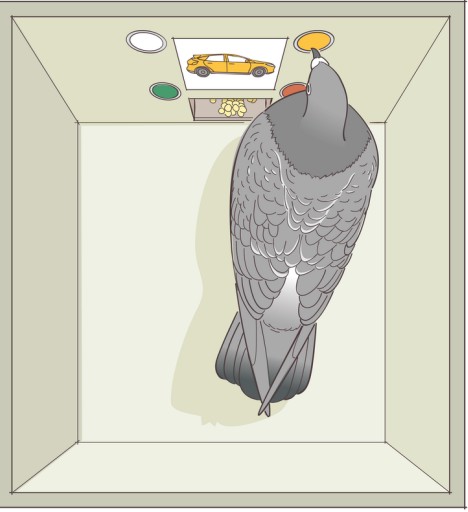
One of the most instructive non-human animal models of drug use disorder is called self-administration. In a self-administration experiment, the subject (usually a monkey, rat, or mouse) gets a surgical implantation of an indwelling intravenous tube that is connected to a pump. Then, the subject is put into an operant conditioning chamber. When they press the lever or poke their nose into a hole, the pump triggers, resulting in an infusion of drug directly into the bloodstream.
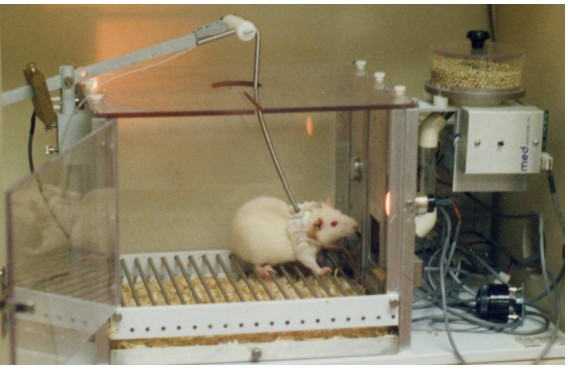
This experimental paradigm closely mimics human drug seeking behavior. For example, rats and mice will very quickly learn to press on the levers for intravenous infusions of cocaine and morphine. Many of them will give themselves drugs even despite severe adverse consequences, such as enduring foot shock – arguably similar to human behavioral patterns where addicts may do all kinds of behaviors for access to drug. Monkeys, if given the option to self administer for cocaine, must be restricted in the amount of drug they receive: if given unrestricted access, they give themselves so much cocaine that they have seizures and die.
A second experimental test created to study drug abuse is called conditioned place preference (CPP). A CPP test works on the principle that animals learn to associate certain environments with positive feelings, and will prefer to spend significantly more time in those environments. To conduct CPP, a three-room testing chamber is used. This experimental chamber has two large rooms, each with characteristics that are unique to that room. For example, one room may have mesh flooring with dark-colored walls, while the other room may have woodchip flooring with light-colored walls. The two rooms are connected by a smaller, neutral room, and the rooms can be isolated from another by means of a dividing door. To conduct CPP, an animal will be given the drug of interest, then placed into one of the two rooms, where they are free to explore the room, but cannot leave. As a control, at a different time, they will be injected with an innocuous solution and placed into the other room. On test day, the animal will be put into the neutral middle room, and given the opportunity to move freely between the two chambers. If the animal developed positive associations with the room where they experienced the drug effect, then they will choose to spend significantly more time in that room. On the other hand, if they experienced negative feelings associated with that room, they will avoid being in the room. Whereas self-administration assesses “drug seeking,” CPP measures “drug liking.”

Below are three major biology-driven hypotheses that have been proposed to explain drug addiction. It’s important to note that none of them tell the complete story – otherwise drug addiction will have already been cured! Also, these theories are not mutually exclusive from one another. A likely answer to curing addiction probably incorporates some combination of these approaches in addition to societal policy changes.
The “hedonia hypothesis”
What drives addicts to behave in the way they do? One of the earlier theories put forward to explain addiction is the hedonia hypothesis proposed in the late 1970s by Roy Wise. The hedonia hypothesis hinges on the assumption that dopamine is the “pleasure neurotransmitter,” and that any substance or behavior that increases dopamine will be desirable. The theory is supported by some behavioral data. For example, using microdialysis, it was found that dopamine release by the mesolimbic pathway increases in the presence of many drugs of abuse. Additionally, administration of dopamine antagonists are able to block some drug taking behavior. This dopamine-centric approach to drug use disorder was so influential that pharmaceutical companies, while developing novel antidepressants, quickly dismissed drugs that increased dopamine out of fear of the risk of addiction potential.
Despite the attractiveness of the straightforward “pleasure neurotransmitter” model, the dopamine hypothesis does not give us the whole picture. For one, dopamine release at the mesolimbic pathway is only notable for psychostimulant drugs, such as cocaine and methamphetamine. But there are plenty of stimulants that still produce the significant overflow of dopamine without causing any rewarding sensation. Furthermore, if dopamine is the signal that mediates the positive hedonic feelings, one might predict that by blocking dopamine receptors chronically, you can also prevent long-term cocaine addiction, but this therapeutic strategy isn’t very useful. Many of the most popular misconceptions about dopamine that you frequently see in the media take this early hypothesis as the truth. It even was featured as the cover of a 1997 Time magazine. That same year in an interview with the original proponent of the hedonia hypothesis published in Science, Roy Wise agreed with recent results that his model was incomplete.
The incentive sensitization model
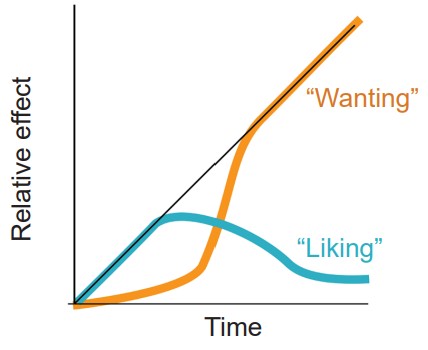
A more recent theory to explain drug addiction is a model developed by researchers from the University of Michigan, Terry Robinson and Kent Berridge. Under their model of drug use, initially a person takes a drug because they like the positive, euphoria-producing side effects of the substance. But after chronic exposure to the substance, the person becomes fixated on obtaining the drug rather than how the drug makes them feel. At this point, they continue to use the drug even though they no longer enjoy the drug to the same degree that they once did. Instead of “liking” the drug, the addict fixates on “wanting” the drug.
The incentive sensitization model suggests that the drug-exposed brain develops an increased sensitivity to drugs, but also more importantly, drug related cues. Pavlovian learning takes place when an addict takes a drug. Each drug has an associated set of cues, and reexposure to those cues can trigger relapse. Over repeated pairings, those cues trigger a progressively stronger response.
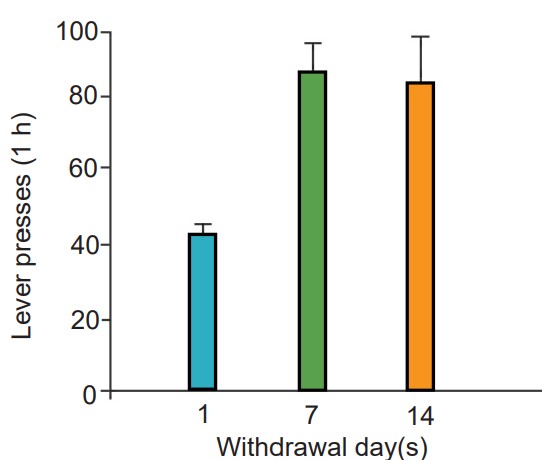
Their model accounts for why it is difficult to remain abstinent from an addictive drug for long durations of time. Despite a decrease in drug seeking behaviors, many addicts eventually relapse, or return to the previous destructive pattern of drug use. One phenomenon that makes staying drug free difficult is the observation that prolonged abstinence can increase the intensity of drug cravings. This is called incubation of craving, and is seen with several different classes of drugs, notably, nicotine.
The Brain Disease Model of Addiction (BDMA)
Much of our current understanding of addiction is based on the principles that repeated drug use leads to biological changes in the brain that resemble aberrant learning. This model for drug addiction is called the Brain Disease Model of Addiction. Genetics has a strong influence on the likelihood that someone develops a disease, and addiction is no different. Inherent risk-taking behaviors, magnitude of a drug effect, and risk of relapse are all modified by genetics.
One of the major implications of the BDMA is that compulsive drug use and seeking is not a choice that a person makes. Addicts are not the way they are because of some poor moral guidance or a weakness of willpower. Addicts compulsively seek drug because of some underlying circuitry of their brain that makes cessation difficult. One substantial accomplishment of the BDMA is that public policy has adjusted to increase access to mental health resources for addicts to help cover the financial challenges of recovery programs (Mental Health Parity and Addiction Equity Act of 2008), and to improve the outcomes of nonviolent drug offenders as they reintegrate with society outside of prisons.
The BDMA is not without fault, however. The major criticism of this framework to explain compulsive drug use is that most people stop their addictive use patterns by themselves spontaneously without any treatment. Another harm of the BDMA is that it puts heavy emphasis on biochemical therapies rather than on a public policy approach to drug addiction treatment. Lastly, the BDMA has not produced a successful therapeutic strategy to help with addiction.
Image credits
11.1 https://pixabay.com/photos/hand-tablets-encapsulate-health-3632914/
11.2 The New Student’s Reference Work, 1914
11.3 https://pixabay.com/photos/syringe-needle-medical-medicine-3902915/
11.4 https://pixabay.com/vectors/lungs-diagram-anatomy-body-human-41562/, https://upload.wikimedia.org/wikipedia/commons/7/7d/Gas_exchange_in_the_aveolus.svg modified by Austin Lim
(Grapefruit) https://pixabay.com/photos/grapefruit-grapefruit-red-citrus-3434196/
11.5 https://upload.wikimedia.org/wikipedia/commons/3/3d/Preoxygenation_before_anesthetic_induction.jpg
11.6 https://upload.wikimedia.org/wikipedia/commons/9/94/Nicoderm.JPG
11.7 https://commons.wikimedia.org/wiki/File:Human_brain_midsagittal_cut.JPG modified by Austin Lim
11.8 https://commons.wikimedia.org/wiki/File:Operant_Conditioning_Involves_Choice.png modified by Austin Lim
11.10 https://commons.wikimedia.org/wiki/File:Scheme_simple_diffusion_in_cell_membrane-ca.svg modified by Austin Lim
11.12 https://commons.wikimedia.org/wiki/File:Scheme_simple_diffusion_in_cell_membrane-ca.svg modified by Austin Lim
11.13 https://commons.wikimedia.org/wiki/File:Richard_M._Nixon_signing_-_NARA_-_194618.tif
11.14 https://pixabay.com/photos/alcoholic-beverages-bar-beer-1845295/
11.15 https://upload.wikimedia.org/wikipedia/commons/0/04/Yeast_%28PSF%29.png
https://pixabay.com/photos/craft-beer-dark-beer-porter-brewery-2468985/
11.16 https://upload.wikimedia.org/wikipedia/commons/4/46/NAchR_2BG9.png
11.17 https://pixabay.com/photos/ecigarette-juul-electronic-cigarette-3576177/
11.18 https://pixabay.com/photos/northern-lights-medical-marijuana-1826032/
11.19 NIH
11.24 Reprinted with consent of Dr. Eugenia Gurevich. https://www.sciencedirect.com/science/article/pii/S0028390817301715?via%3Dihub#undfig1
11.26 https://upload.wikimedia.org/wikipedia/commons/c/c4/SelfAdmin-2.jpg
11.27 https://upload.wikimedia.org/wikipedia/commons/0/0a/Conditioned_Place_Preference_-_Floor_Texture.JPG
Reprinted with consent of Dr. Ewa Niedzielska-Andres
https://www.sciencedirect.com/science/article/pii/S0166432819302803?via%3Dihub
11.29 Reprinted with consent of Dr. Xia Li https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5446935/
a disorder affecting a persons ability to control their use of various substances
the study of drugs that affect the nervous system
The field of study of the effect of drugs on the body
the study of the effects of the body on drugs
ways for drugs to enter the body
absorbed through the gastrointestinal tract
route of administration that avoids the digestive tract
route of administration by mouth
a branch of the circulatory system that surrounds the gastrointestinal tract
causes a decrease in difference between the consumed amount of drug and the amount of drug that gets to its target of action
drugs that are inactive until they get converted into another substance after enzymatic processing
an inactive chemical, but once it is exposed to acidic conditions, it becomes psilocin. found in "magic mushrooms"
route of administration that avoids a majority of the first-pass metabolism
refers to placement of the substance under the tongue
refers to drugs that are absorbed by the vasculature in the gums
drug is delivered straight into the bloodstream. fastest route of administration
drug is delivered into a muscle. slower than IV injections but easier to perform
solution is injected into the fat layer underneath the skin
route of administration where substances are breathed in through the nose or mouth
a class of enzymes in the liver and the small intestines that metabolize a wide variety of substances that enter the body
low blood levels of oxygen
route of administration where the substance is pulverized into a fine powder then inhaled through the nose
substance is placed on the skin surface, and over time the chemical diffuses through the skin into the blood vessels
similar to transdermal, topical administration involves a substance placed on the skin surface, but does not significantly increase the blood concentration of the drug
contains neurons that synthesize the neurotransmitter dopamine
consists of dopamine producing neurons that release dopamine onto the cells in the nucleus accumbens
processing center for reward and motivation
short for nucleus accumbens
another term for nucleus accumbens
projection of prefrontal cortex
the opposite reaction to reward
VTA's antireward pathway
a special cage equipped with a device which the subject can physically manipulate
another name for a operant conditioning chamber
the experimental paradigm of the Skinner Box
describes the randomness of the movement of molecules
a graph that plots the activation of a receptor on the y-axis, and increasing dosage of a drug on the x-axis
Chemicals that bind to the active site of ionotropic receptors
chemical substances that can activate receptors
another term for the active site
The specific series of amino acid residues on the extracellular surface of a receptor where neurotransmitter molecules bind
substance that can activate the receptor to the maximal degree at high concentrations.
substances that can also activate the receptor by binding to the orthosteric site, but are unable to fully activate the receptor, even at increasingly higher doses
causes an opposite response as an agonist
the substances that prevent agonists from acting
substances that bind to the orthosteric site
substances that bind to the allosteric site
found on extracellular side of the receptor and consists of a special three-dimensional arrangement of amino acids
allosteric modulators increase the action of the agonist
short for positive allosteric modulator
allosteric modulators decrease the action of the agonist
negative allosteric modulator
drugs with many sites of action
chemicals that can act on the nervous system which can induce a change in behavior or mindstate
easily-obtained substance with a highly intoxicating effect
ionotropic receptors of the nervous system
a drug that is derived from the flowering buds or other parts of the Cannabis sativa plant
the main psychoactive ingredient in cannabis
shirt for delta-9-tetrohydrocannabinol
receptors that our body uses endogenously
the first type of cannabinoid receptor. exerts the majority of the psychoactive effect
second type of cannabinoid receptor. works through the immune system
substances our bodies naturally produce that activate cannabinoid receptors
short for endocannabinoids
inhibitory metabotropic receptors which signal using the Gαi protein
the most well known natural opioid
describes the dramatic increase in the number of opioid related overdose deaths
substances that activate the sympathetic nervous system
large membrane-spanning proteins that function to move molecules from the extracellular space back into the presynaptic terminal
increases the concentration of neurotransmitters in the synapse
a manmade psychedelic
occurs when the body is better at eliminating a drug, so more of the drug is needed to feel the effects
another term for metabolic tolerance
another term for metabolic tolerance
characterized by a decrease in receptor expression after chronic exposure to an agonist
another term for functional tolerance
a form of tolerance that is dependent on Pavolvian cues
a phenomenon opposite to tolerance: instead of a drug effect being lessened after chronic exposure, a sensitized person experiences an increase in drug effect
a set of symptoms that a person experiences when they are abstinent from the substance
having the urge to take the drug when someone is in withdraw
an experiment where subject animals receive drugs via infusion when they perform certain actions
experimental test that works on the principle that animals learn to associate certain environments with positive feelings, and will prefer to spend significantly more time in those environments.
hinges on the assumption that dopamine is the “pleasure neurotransmitter,” and that any substance or behavior that increases dopamine will be desirable.
observation that prolonged abstinence can increase the intensity of drug cravings
the principle that repeated drug use leads to biological changes in the brain that resemble aberrant learning
